Zoran Markovic*
Chemistry Department, Faculty of Science, University of Kragujevac;
Bogdan Solaja, Dragana Milic, Ivan Juranic*
Faculty of Chemistry, University of Belgrade, 11001 Belgrade, P.O.B. 158
Miroslav Gasic
Institute of Chemistry, Technology and Metallurgy, Belgrade, Yugoslavia
Summary. - The MO study has shown that the radical oxidation of phenols into quinols occurs
readily. Further radical oxidation (with m-CPBA / (BzO)2 / hNU system) of quinols occurs
through appropriate biradical species with the activation energy of 19 kcal/mol yielding
syn-epoxyquinols. The stereochemical outcome presented in this study is in full agreement with
the experimental results.
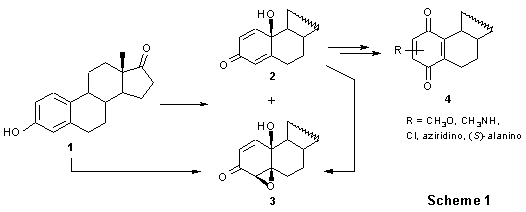
Introduction. - During the synthesis of steroidal quinones 4 (Scheme 1) it was found that phenols 1 can be transformed into quinols 2 and epoxyquinols 3 using m-chloroperoxybenzoic acid (m-CPBA) as an oxidant.1,2 It has been discovered that the transformations 1 2 + 3 occur by radical mechanism yielding m-chlorobenzoic acid in 96% yield (on 15 g scale).1 The epoxidation reaction 2 3 has been also found to be a radical one since it does not occur without an initiator and stops in O2 atmosphere.
In order to provide further support to our findings, the results of MO study of phenol-to-quinol
and quinol-to-epoxyquinol transformations are presented.
Method of calculation. - The structures of compounds were built by PC MODEL, version 4.0,3 that involves an MMX force field,4 and were saved as MOPAC files for PM3 semiempirical calculations.5
We used the MNDO-PM3 method that proved to be highly reliable for investigating molecular
properties of molecules and radicals,6,7 and MOPAC program package, version 7.01. The
geometries of all molecular species correspond to energy minima in vacuum, and were optimized
by the PM3 method. The transition states for all reactions were located using corresponding
MOPAC facilities (TS, SADDLE). Obtained structures were refined with NLLSQ when needed,
and transition states were proved by vibrational analysis showing only one negative vibration.
Results and Discussion
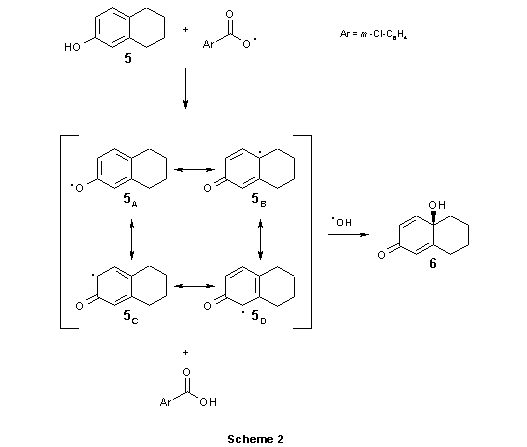
In order to make the most detailed correlation with experimental results we used the corresponding 6-hydroxy-1,2,3,4-tetrahydronaphtalene 5, instead of steroidal phenol (Scheme 2). Both radicals, OH and m-Cl-C6H4COO, participating in this reaction were generated from meta-chloroperoxybenzoic acid (m-CPBA) under influence of radical initiator, benzoyl peroxide, and light.1,2,(1)
The extensive calculations were performed by means of MNDO-PM3 method, involving starting
compounds, all products and radical intermediates, given in Schemes 2 and 3. The energies of the
most stable conformations of starting compound 5, intermediates 5A-D and 6A-C, as well as
products 6, 7 and 8, are presented in Table 1.
| Table 1. Heats of formation for products and intermediates [kcal/mol] | |||||||
| molecular species | Hf | molecular species | Hf | molecular species | Hf | molecular species | Hf |
| 5 | -40.9 | 6A | -98.1 | 6D | 91.6 | 7 | -79.1 |
| 5A-D | -17.3 | 6B | -95.0 | 6E | 53.9 | 8 | -76.8 |
| 6 | -61.6 | 6C | -96.9 | 6F | 15.2 | ||
In the first step the reactive species, m-Cl-C6H4COO radical abstracts the phenol hydrogen atom (ArO-H) giving corresponding radical 5A-D. This radical can be described as a superposition (linear combination) of four wave functions. On the basis of the spin densitiy distribution, the greatest weight has the wave function 5B. Calculation of the reaction trajectory for the attack of m-Cl-C6H4COO on phenol group of compound 5 does not reveal any transition state. The formation of intermediate 5A-D is an exothermic process, and no activation energy is needed. Based on this finding we can conclude that the first step of the reaction is very fast.
Next step of this reaction, the approach of two radical moieties (5A-D and OH), proceeds
smoothly giving the product 6. Calculation of a reaction trajectory for this attack does not reveal
any transition state. Because of the larger steric congestion at the ![]() -side, the formation of
compound 6 from radical 5A-D, by the approach to
-side, the formation of
compound 6 from radical 5A-D, by the approach to ![]() -side, is thermodynamically strongly
favoured.
-side, is thermodynamically strongly
favoured.
For the next step it was assumed that OH radical is reactive species which attacks the C4-C5
double bond(2) in compound 6. Carbons C4 and C5 can be attacked from either ![]() - or
- or
![]() -side. The approach of two reacting species would proceed giving intermediates 6A-6D
(Scheme 3).
-side. The approach of two reacting species would proceed giving intermediates 6A-6D
(Scheme 3).
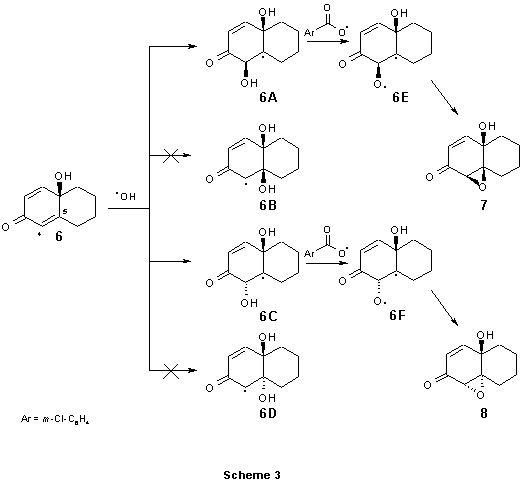
As expected, our calculations showed that attack at C5 is unfavourable for two reasons. Access
to C5 from ![]() -side is impossible because of steric bulk near the reactive site. For this
reason intermediate 6D is not further considered. The approach to
-side is impossible because of steric bulk near the reactive site. For this
reason intermediate 6D is not further considered. The approach to ![]() -side (intermediate
6B) is energetically less favoured as compared to 6A and 6C and it is higher in energy by 3.1 and
1.9 kcal/mol, respectively. For this reason, the intermediate 6B is also excluded from the
discussion below.
-side (intermediate
6B) is energetically less favoured as compared to 6A and 6C and it is higher in energy by 3.1 and
1.9 kcal/mol, respectively. For this reason, the intermediate 6B is also excluded from the
discussion below.
The arguments presented above indicate that the approach of OH radical to C4 is preferred to
the C5 attack. Calculations also show that the ![]() -attack to C4 is favoured over
-attack to C4 is favoured over ![]() -attack by 1.2 kcal/mol, corresponding to 6.4 times faster formation of 4
-attack by 1.2 kcal/mol, corresponding to 6.4 times faster formation of 4![]() -intermediate
6A. This calculation indicates the formation of
-intermediate
6A. This calculation indicates the formation of ![]() -epoxy compound 7 as major product
what is in good agreement with experimental data.1,2
-epoxy compound 7 as major product
what is in good agreement with experimental data.1,2
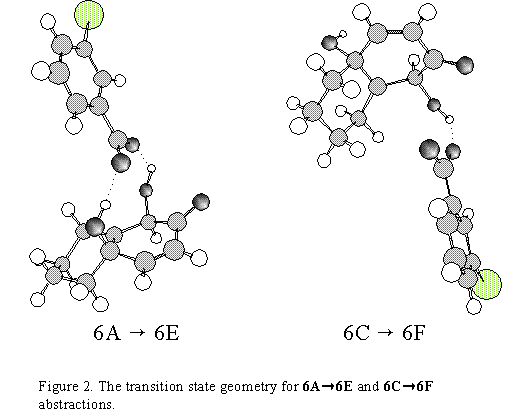
The final step is a ring closure reaction of 6A(7) or 6C(8). Two possibilities were anticipated
(Scheme 3): a) H-OC(4) abstraction by m-Cl-C6H4COO radical followed by collapse of the
formed biradical 6E. b) Formation of protonated epoxide (7H) followed by hydrogen
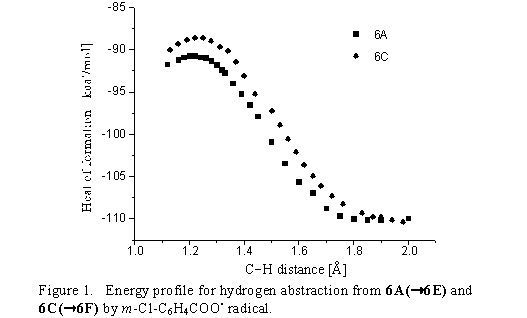
abstraction. It was found that the corresponding transition states differ by 36 kcal/mol: a) Ea6A6E
= 19 kcal/mol, b) Ea6A7H = 55 kcal / mol (structure 7H is actually transition state for the
conversion 6A to 6B) and consequently, we propose that epoxyquinol oxirane ring formation
(from quinol, by m-CPBA) proceeds through biradicals 6E or 6F.(3) In addition, the isolation of
only the cis-isomer of epoxyquinol is also driven by surplus activation energy difference of 2.6
kcal/mol in favour of 6A 6E 7 process (Ea6A6E = 19 kcal/mol; Ea6C6F = 21.6 kcal/mol).
The Ea of 2.6 kcal/mol (Figure 1) taken together with preferred formation of 6A as compared to
6C, is in good agreement with experimental results. In Figure 2 the transition states of two
reaction pathways are shown. It is obvious that ![]() -approach is facilitated by hydrogen
bonding with C(10)-ALPHA-OH group.
-approach is facilitated by hydrogen
bonding with C(10)-ALPHA-OH group.
Conclusion
In the presented MO study was shown that the radical oxidation of phenols to quinols (5 6) is a favoured reaction pathway. The radical epoxidation reaction of quinol 6 occurs through appropriate biradical species with the activation energy of 19 kcal/mol, yielding syn-epoxyquinol 7.
The stereochemical implications resulting from this study are in full agreement with the
experimental results. Corollary, the MO calculations confirm that free-radical mechanism is a
reasonable explanation of experimental findings.
REFERENCES
1. B.A. Solaja, D. R. Milic, and M.J. Gasic, Tetrahedron Lett. 1996, 37, 3765-3768.
2. D.R. Milic, M.J. Gasic, W. Muster, J.J. Csanádi, and B.A. Solaja, Tetrahedron 1997, 53, 14073-14084.
3. Serene Software Box Bloomington IN 45402-3076
4. (a) K. E. Gajevski, K.E. Gilbert, and J. McKelvey, Adv. Mol. Mod., 1990, 2, 65; (b) U. Burket, N. L. Allinger, Molecular Mechanics, American Chemical Society, Washington, D.C. 1982.
5. For PM3 parametrization of MNDO see: (a) J.J.P. Stewart, J. Comp. Chem., 1989, 10, 109; (b) J. J. P. Stewart, J. Comp. Chem., 1989, 10, 221.
6 (a) J. J. P. Stewart, J. Comp. -Aided Molec. Des., 1990, 4(1), 1; (b) J. J. P. Stewart, QCPE # 455.
7. (a) I.O. Juranic, M.Lj. Mihailovic and M. Dabovic, J. Chem. Soc., Perkin Trans. 2, 1994,
872-881; (b) R.C. Bingham, M.J.S. Dewar and D.H. Lo, J. Am. Chem. Soc., 1975, 97, 1285; (c)
P. Bischof, Croat. Chem. Acta, 1980, 53, 51; (d) M.J.S. Dewar, G. Ford, H.S. Rzepa and Y.
Yamaguchi, J. Mol. Struct., 1978, 43, 1325; (e) H.S. Rzepa and W.A. Wylie, J. Chem. Soc.,
Perkin Trans. 2, 1991, 939.
1. It has been confirmed that m-CPBA decomposes much faster (ca. six times) in the presence of 10 % (BzO)2 under reaction conditions applied.
2. Steroid numbering
3. No activation energy for the collapse of biradical.