Pblished in: J. Serb. Chem. Soc., 1998, 63(5), 359-365.
Reactivity of ![]() -aroylacrylic acids with diazodiphenylmethane
-aroylacrylic acids with diazodiphenylmethane
Lj. S. Stevovic1, B. J. Drakulic2, I. O. Juranic3, S.Z. Drmanic4 and B.Z. Jovanovic4
1INOTEH-Belgrade; 2 Center for Chemistry IChTM, Belgrade; 3 Faculty of Chemistry,
University of Belgrade; 4Faculty of Technology and Metallurgy, University of Belgrade,
Yugoslavia
Abstract
The rate constants for 10 trans-substituted ![]() -aroylacrylic acids in their reaction with
diazodiphenylmethane (DDM) were determined in ethanol at 30 oC. The intention was to study
the transmission of electronic effects of substituents on the phenyl nuclei to the carboxylic group
reaction center, through the C(=O)CH=CH group. The obtained rate constants were
analyzed with the classical Hammett equation and the extended Hammett and Taft equation. It
was possible, using Taft's relationship, to effect the separation of the overall polar effect into
inductive and resonance components. The pattern of transmission through the observed
conjugated system has been discussed. Conformational variations caused by variable
substituents in an aromatic ring were calculated using the MNDO-AM1 semiempirical method.
The inclusion of a calculated conformational effect considerably improves the regression.
-aroylacrylic acids in their reaction with
diazodiphenylmethane (DDM) were determined in ethanol at 30 oC. The intention was to study
the transmission of electronic effects of substituents on the phenyl nuclei to the carboxylic group
reaction center, through the C(=O)CH=CH group. The obtained rate constants were
analyzed with the classical Hammett equation and the extended Hammett and Taft equation. It
was possible, using Taft's relationship, to effect the separation of the overall polar effect into
inductive and resonance components. The pattern of transmission through the observed
conjugated system has been discussed. Conformational variations caused by variable
substituents in an aromatic ring were calculated using the MNDO-AM1 semiempirical method.
The inclusion of a calculated conformational effect considerably improves the regression.
Introduction
Diazodiphenylmethane (DDM) reacts with carboxylic acids as follows:
RCO2H + Ph2C=N2 -> RCO2CHPh2 + N2
In alcoholic solutions a concurrent reaction takes place with the alcohol, catalyzed by the acid.
ROH + Ph2C=N2 -> ROCHPh2 + N2
For the reaction in alcoholic media Bowden et al.1 and O'Ferrall et al.2 concluded that the two reactions have a common rate-determining step, viz. proton transfer from the acid to the diazodiphenylmethane.
This reaction is one of most frequently used methods for the study of substituent effects in
carboxylic acid reactivity investigations. It was successfully applied for the determination of rate
constants and their subsequent analysis, using the linear free energy relationship, in a series of
substituted benzoic3,4, phenylacetic5, cinnamic6,7, pyridine and pyridine-N-oxide8, ![]() -phenyl
pyridine acrylic acid9. In these investigations, the quantitative effects of the substituent on the
reactivity of the carboxylic group reaction centers were analyzed.
-phenyl
pyridine acrylic acid9. In these investigations, the quantitative effects of the substituent on the
reactivity of the carboxylic group reaction centers were analyzed.
In the present work, a series of substituted trans-![]() -aroylacrylic acids of the general formula:
-aroylacrylic acids of the general formula:
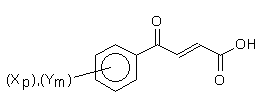
Where: X, Y: H, H (1); Me, H (2); Et, H (3); i-Pr, H (4); t-Bu, H (5); H, NO2 (6); OCH3, H (7);
Cl, Cl (8); Me, NO2 (9); CH3, CH3(10).
were synthesized and their reactivity investigated.
Results and discussion
In Table I, the rate constants for the reaction of the investigated trans-![]() -aroylacrylic acids with
DDM, experimentally determined in ethanol at 30 oC, are given. In the same Table the
corresponding
-aroylacrylic acids with
DDM, experimentally determined in ethanol at 30 oC, are given. In the same Table the
corresponding ![]() m and
m and ![]() p constants, used for analysis of the substituent effects, are also presented.
p constants, used for analysis of the substituent effects, are also presented.
Table I. Rate constants for the reaction of trans-![]() -aroylacrylic acids with DDM in ethanol at 30
oC, and appropriate substituent constants.
-aroylacrylic acids with DDM in ethanol at 30
oC, and appropriate substituent constants.
| Acid | k
(dm3 mol-1 s-1) |
||
| 1 | 0.088 | 0 | 0 |
| 2 | 0.0785 | 0 | -0.17 |
| 3 | 0.082 | 0 | -0.15 |
| 4 | 0.0815 | 0 | -0.15 |
| 5 | 0.079 | 0 | -0.20 |
| 6 | 0.095 | 0.71 | 0 |
| 7 | 0.054 | 0 | -0.27 |
| 8 | 0.107 | 0.37 | 0.23 |
| 9 | 0.086 | 0.71 | -0.17 |
| 10 | 0.075 | -0.07 | -0.17 |
* ![]() m and
m and ![]() p values are from Ref. (10)
p values are from Ref. (10)
The use of the classical Hammett equation (1)
log k = ![]() (
(![]() m +
m + ![]() p ) + log k0 (1)
p ) + log k0 (1)
where k0 is the rate constant of the basic member of the series, ![]() -aroylacrylic acid, k the reaction
constants of the substituted acids (2-10),
-aroylacrylic acid, k the reaction
constants of the substituted acids (2-10), ![]() the reaction constant,
the reaction constant, ![]() m and
m and ![]() p, the substituent
constants of acids with substituents in m- and p-position of the phenyl nucleus, gave an
unsatisfactory correlation (2):
p, the substituent
constants of acids with substituents in m- and p-position of the phenyl nucleus, gave an
unsatisfactory correlation (2):
log k = 0.2082 (![]() m +
m + ![]() p) -1.0875 (2)
p) -1.0875 (2)
(n=10; s=0.0440; r=0.8583)
The low correlation coefficient ( r) shows that, in the observed series, the effects of the substituents are more complex, and are not well interpreted by the simple Hammett correlation (1).
The use of the extended Hammett equation, of the form (3)
log k = ![]() m
m ![]() m +
m + ![]() p
p ![]() p + log k0 (3)
p + log k0 (3)
gives a better insight into the substituent effects, and it was possible to distinguish the effects in the m- and p-position of the phenyl nucleus:
log k = 0.163 ![]() m + 0.340
m + 0.340![]() p -1.0659 (4)
p -1.0659 (4)
( n=10; s=0.0310; r=0.8807)
The almost satisfactory correlation obtained cannot be trusted because of the disproportion
between ![]() m and
m and ![]() p. The improved correlation is an obvious artefact making the substituents in
the m-position exert a minor influence.
p. The improved correlation is an obvious artefact making the substituents in
the m-position exert a minor influence.
Analysis of the nature of the substituents in the p-position of the phenyl nucleus shows that the
majority are electron donors, the OCH3 group being an exceptionally good electron donor, and
evidently strongly influences the overall correlation. It is not surprising, therefore, that
exceptionally good results from the extended Hammett equation were obtained if the ![]() p+ value
were used, which account for the direct conjugation of the oxygen lone pair from the methoxy
group with the carboxylic reaction center.
p+ value
were used, which account for the direct conjugation of the oxygen lone pair from the methoxy
group with the carboxylic reaction center.
The result was as follows:
log k = 0.0446 ![]() m + 0.2725
m + 0.2725![]() p+ -1.0310 (5)
p+ -1.0310 (5)
( n=10; s=0.0093; r=0.9838; for p-OCH3 and p-Cl from Ref. (10))
The comment on the value of the correlation coefficient is the same as for Eq. 4.
A remarkable correlation was obtained using the Taft equation (6)
log k = ![]() I
I ![]() I +
I + ![]() R
R ![]() R + log k0 (6)
R + log k0 (6)
whereby it was possible to dissect the overall substituent effect into an inductive and a resonance component.
If the rate data for the set of 10 substituted acids (Table I), were correlated using Eq. (6) the following parameters were calculated:
log k = 0.1693 (![]() I + 2.795 R
I + 2.795 R![]() ) -1.0326 (7)
) -1.0326 (7)
( n=10; s=0.0098; r=0.9869)
(The values for ![]() I and
I and ![]() R are: 0.47 and -0.25 for -Cl; -0.01 and -0.16 for -CH3; -0.01 and
-0.014 for -C2H5; 0.01 and -0.16 for -CH(CH3)2; 0.01 and 0.18 for -C(CH3)3; 0.67 and 0.10
for NO2; and 0.30 and -0.58 for -OCH3. The optimized
R are: 0.47 and -0.25 for -Cl; -0.01 and -0.16 for -CH3; -0.01 and
-0.014 for -C2H5; 0.01 and -0.16 for -CH(CH3)2; 0.01 and 0.18 for -C(CH3)3; 0.67 and 0.10
for NO2; and 0.30 and -0.58 for -OCH3. The optimized ![]() coefficient for
coefficient for ![]() m=
m=![]() I+
I+![]()
![]() R is
-0.098)18
R is
-0.098)18
The negative ![]() value for the resonance contribution to
value for the resonance contribution to ![]() m is hard to explain.
m is hard to explain.
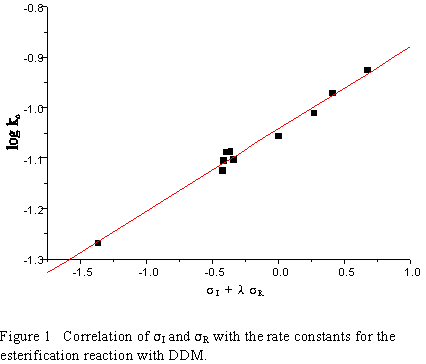
A much better correlation is obtained with ![]() +R values (see Fig. 1):
+R values (see Fig. 1):
log kc = 0.1641(![]() I + 2.515
I + 2.515 ![]() +R) -1.0415 (8)
+R) -1.0415 (8)
( n=10; s=0.0070; r=0.9929)
(The values for ![]() R+ are: -0.21 for -Cl; -0.16 for -CH3; -0.014 for -C2H5; -0.16 for -CH(CH3)2; -0.13 for -C(CH3)3; 0.10 for NO2; and -0.66 for -OCH3. The optimized
R+ are: -0.21 for -Cl; -0.16 for -CH3; -0.014 for -C2H5; -0.16 for -CH(CH3)2; -0.13 for -C(CH3)3; 0.10 for NO2; and -0.66 for -OCH3. The optimized ![]() coefficient for
coefficient for ![]() m=
m=![]() I+
I+ ![]()
![]() R is 0.0)18
R is 0.0)18
This good correlation has another important feature. The constant (-1.0415) has value very close
to log kH = -1.0555 making the other coefficients physically reliable. The ![]() factor for the meta
substituents is zero. The low value for
factor for the meta
substituents is zero. The low value for ![]() is accountable on the basis of AM1 calculations, which
reveal that all aroylacrylic acids have the benzene ring rotated at a dihedral angle of 20-30 to the
double bond plane.
is accountable on the basis of AM1 calculations, which
reveal that all aroylacrylic acids have the benzene ring rotated at a dihedral angle of 20-30 to the
double bond plane.
A further improvement in the correlation is obtained by including the effect of a variable
torsional angle between the aromatic ring and the side chain. The torsional angles are listed in
Table II.
Table II. Torsional angles of the aromatic ring in aroylacrylic acids for various substituents.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| X,Y | H | p-Me | p-Et | i-Pr | t-Bu | m-NO2 | p-OCH3 | p-Cl,
m-Cl |
p-Me,
m-NO2 |
p-CH3
m-CH3 |
| |
37.28 | 37.88 | 37.88 | 37.90 | 37.14 | 35.67 | 39.64 | 36.17 | 36.00 | 37.87 |
The result of the correlation with ![]() I and
I and ![]() R (see comment for Eq. 7) is
R (see comment for Eq. 7) is
log k = 0.3730 (![]() I + 2.535
I + 2.535 ![]() R) + 2.2345 - 5.1282 cos2
R) + 2.2345 - 5.1282 cos2![]() (9)
(9)
( n=10; s=0.0034; r=0.9915); calculated log kH = -1.0122
The result of the correlation with ![]() I and
I and ![]() R+ (see comment for Eq. 8) is
R+ (see comment for Eq. 8) is
log k = 0.3252 (![]() I + 2.305
I + 2.305 ![]() R) + 1.6055 - 4.1667 cos2
R) + 1.6055 - 4.1667 cos2![]() (10)
(10)
( n=10; s=0.0021; r=0.9971); calculated log kH = -1.0324
The values for ![]() are the same as in (7) and (8).
are the same as in (7) and (8).
From Table II it can be seen that all the torsional angles are in the range between 35 and 45.
Nevertheless, the variations conform to the electronic demand of the reaction, and a considerable
improvement of the correlation is obtained. The small interval of the ![]() values is balanced by the
large coefficients for cos2
values is balanced by the
large coefficients for cos2![]() in equations (9) and (10).
in equations (9) and (10).
Experimental
Rate measurements. The rate constants for the reaction of DDM with p-substituted ![]() -aroylacrylic acids were determined by the spectroscopic method proposed by Roberts and his co-workers.11 The absorbancy measurements were performed at 30 oC in ethanol at 525 nm using 1
cm cell. A SHIMADZU UV 160A spectrophotometer was used. The second order rate constants
for all the investigated compounds were obtained by dividing the pseudo first order rate constants
by the acid concentration. The concentration of acid was 0.06 M and of DDM 0.006 M.
-aroylacrylic acids were determined by the spectroscopic method proposed by Roberts and his co-workers.11 The absorbancy measurements were performed at 30 oC in ethanol at 525 nm using 1
cm cell. A SHIMADZU UV 160A spectrophotometer was used. The second order rate constants
for all the investigated compounds were obtained by dividing the pseudo first order rate constants
by the acid concentration. The concentration of acid was 0.06 M and of DDM 0.006 M.
Materials. DDM was prepared by the Smith and Howard method12. Stock solutions (c.a. 0.06 M) were stored in a refrigerator and diluted for use. The ethanol was UV-spectroscopic grade.
Method of calculation. The geometries and charge distributions of the molecules were determined by the AM1 method (using a MOPAC package, Version 7.01),13 employing full geometry optimization and imposing no a priori symmetry constraints. The MNDO-AM1 method was proven to be accurate for the calculation of various molecular species.14
Synthesis of aroylacrylic acids: Trans-![]() -aroylacrylic acids were prepared, according to the Papa
et al.15 by a modification of the Friedel-Crafts reaction, by adding an aromatic substrate to a
solution of maleic anhydride and anhydrous aluminum trichloride (molar ratio 1:2) in 1,1,2,2-tetrachloroethane. Instead of tetrachloroethane we used 1,2-dichloroethane and moderately higher
yields were obtained. The only exception was trans-
-aroylacrylic acids were prepared, according to the Papa
et al.15 by a modification of the Friedel-Crafts reaction, by adding an aromatic substrate to a
solution of maleic anhydride and anhydrous aluminum trichloride (molar ratio 1:2) in 1,1,2,2-tetrachloroethane. Instead of tetrachloroethane we used 1,2-dichloroethane and moderately higher
yields were obtained. The only exception was trans-![]() -benzoylacrylic acid that was prepared
according to Grummit et al.16 trans-
-benzoylacrylic acid that was prepared
according to Grummit et al.16 trans-![]() -(3-Nitrobenzoyl)acrylic acid and trans-
-(3-Nitrobenzoyl)acrylic acid and trans-![]() -(3-nitro-4-methylbenzoyl)acrylic acid were prepared by nitration of the mother acids in fuming nitric acid at
0 oC, according to Bogert and Ritter.17
-(3-nitro-4-methylbenzoyl)acrylic acid were prepared by nitration of the mother acids in fuming nitric acid at
0 oC, according to Bogert and Ritter.17
Typical experimental procedure
In a 100 ml two-necked flask equipped with magnetic stirrer, reflux condenser and dropping funnel, 6.125 g (62.5 mmol) of maleic anhydride was suspended in 25 ml of dry 1,2-dichloroethane. After 10 minutes 15.5 g (125 mmol) of powdered anhydrous aluminum trichloride was added and the reaction mixture was stirred for another 20 minutes, until a homogeneous yellow suspension was formed. An aromatic substrate (62.5 m mol) was added at such a rate to keep the temperature (below 50 oC) and foaming under control. The reaction mixture was stirred for 9 h at 20 oC; 0.5 h at 60 oC; refluxed for another 0.5 h and then poured into 200 g of ice/water mixture (1:1) with 20 ml concentrated hydrochloric acid. The dichloroethane was removed by steam distillation. The crude acid was collected by filtration, dissolved at 20 oC in water with sodium carbonate at pH 8.5-9.0 and traces of aluminum hydroxide were filtered off. The mother liquor was acidified with hydrochloric acid to pH 1.0 and the pure acid was collected by filtration, washed with water and dried in the open air. Yields, recrystallization solvents and melting points are given in Table III.
Table III. Crystallisation solvent, melting points and yields* of compounds (1-10)
| Acid | Solvent | M.p. oC | Yield % |
| 1 | H2O | 160 | 93 |
| 2 | Benzene | 139 | 85 |
| 3 | Benzene | 106 | 94.5 |
| 4 | Benzene | 103 | 73 |
| 5 | Benzene | 125 | 75 |
| 6 | C2H5OH | 193-195 | 85 |
| 7 | Benzene | 139 | 72 |
| 8 | Benzene | 143 | 66 |
| 9 | C2H5OH | 178-180 | 90 |
| 10 | C2H5OH | 123 | 75 |
* Compare the data in reference 15 and in: Ernst Berliner, Chapter 5, in Roger Adams Ed.,
"Organic Reactions". J.Wiley, London 1949. Vol. 5, p. 285.
References
1. K. Bowden, A. Buckley, N.B. Chapman, and J. Shorter, J. Chem. Soc., (1964) 3380.
2. R.A. More O'Ferrall, W.K. Kwok and S.I. Miller, J. Am. Chem. Soc., 86 (1964) 5553.
3. A. Buckley, N. B. Chapman, M. R. J. Dark, J. Shorter and H. M. Wall, J. Chem. Soc. (B), (1968) 631.
4. M. H. Aslam, N. B. Chapman, J. Shorter and M. Charton, J. Chem. Soc., Perkin II, (1981) 720.
5. N. B. Chapman, J. R. Lee and J. Shorter, J. Chem Soc. (B), (1969) 769.
6. 3. N. B. Chapman, D. J. Newman, J. Shorter and H. M. Wall J. Chem Soc. (B), (1976) 847.
7. G. Uscumlic, V. Krstic and M. Muskatirovic, Quant. Struct. Act. Relat. 10 (1991) 216.
8. Dj. Dimitrijevic, Z. Tadic, M. Misic-Vukovic and M. Muskatirovic, J. Chem Soc. Perkin II, (1974) 1051; M. Radojkovic-Velickovic and M. Misic-Vukovic J. Chem Soc. Perkin II, (1984) 1975.
9. M. Misic-Vukovic, S. Drmanic and B. Jovanovic Heterocycles, 37 (1994) 1503.
10. R. A. Y. Jones, "Physical and mechanistic organic chemistry", Cambridge University Press, Cambridge, 1979, p. 55.
11. J. D. Roberts, M. A. Mc Elhil and R. Armstrong, J. Am. Chem. Soc., 71 (1949) 2923.
12. L. I. Smith and K. L. Howard, Org. Synth., Coll. Vol III, (1955) 351.
13. J.J.P Stewart, .J. Comput. Chemistry, 2 (1989) 221-264.
14. T. Wyttenbach, G. Vonhelden, MT. Bowers, J. Am. Chem. Soc. 118 (1996) 8355-8364; H. Bock, S. Nick, Seitz, W., C. Nather, JW. Bats, Z, Naturforsch. B - J. Chem. Sci. 51 (1996) 153-171; GH. Peslherbe, HB. Wang, WL. Hase, J. Am. Chem. Soc. 118 (1996) 2257-2266; WP. Hu, DG. Truhlar, J. Am. Chem. Soc. 118 (1996) 860-869.
15. D. Papa, F. Swenk, F. Villani and E. Klinsberg, J. Am. Chem. Soc. 70 (1948) 3356.
16.O. Grummit, E. I. Backer and C. Miesse "Organic Syntheses" Coll. Vol. III, (1955) 108.
17. M.T. Bogert and J.J. Ritter, J. Am. Chem. Soc. 47 (1925) 532.
18. M. Charton, in R.W. Taft, Ed. Progress in Physical Organic Chemistry, Vol. 13. New York 1981, pp. 119-252.