Ljubinka Lorenc, Vladimir Pavlovic, Ivan Juranic(2) and Mihailo Lj. Mihailovic
Faculty of Chemistry, University of Belgrade, Studentski trg 12-16, P. O. Box 158, YU-11001
Belgrade, Yugoslavia,
Lidija Bondarenko-Gheorghiu, Natalija Krstic and Milan Dabovic,
Center for Chemistry, ICTM, P. O. Box 815, YU-11001 Belgrade, Yugoslavia
Summary: - The thermal and acid-catalyzed intramolecular rearrangement of the (Z)- and (E)-cyclodecene-1,4-dione compounds deriving from steroids, 2a,b and 3a,b, respectively, proceeds
stereoselectively to give the corresponding configurationally different spiro-![]() -lactone
derivatives, the (5R,9R)-isomers 4a,b (from the (Z)-cyclodecenediones 2a,b) and the (5R,9S)-isomers 5a,b (from the (E)-cyclodecenediones 3a,b). The semiempirical MNDO-AM1
molecular-orbital method was applied to elucidate the possible mechanistic pathway of the
observed intramolecular process leading to the spiro-
-lactone
derivatives, the (5R,9R)-isomers 4a,b (from the (Z)-cyclodecenediones 2a,b) and the (5R,9S)-isomers 5a,b (from the (E)-cyclodecenediones 3a,b). The semiempirical MNDO-AM1
molecular-orbital method was applied to elucidate the possible mechanistic pathway of the
observed intramolecular process leading to the spiro-![]() -lactone structures.
-lactone structures.
Introduction
Recently it was reported1,2 that the stereoisomeric (Z)- and (E)-6,9-dioxocyclodec-3-enyl
derivatives 2a,b and 3a,b, respectively (Scheme 1) (obtained by oxidative fragmentation of the
C(5)C(10) bond in 5-hydroxy-8-oxo-8,14-seco-5![]() -androstane-3
-androstane-3![]() ,17
,17![]() -diyl
diacetate (1a) (X=H2, R=OAc),3,4 and 5-hydroxy-8,14-dioxo-8,14-seco-5
-diyl
diacetate (1a) (X=H2, R=OAc),3,4 and 5-hydroxy-8,14-dioxo-8,14-seco-5![]() -cholestane-3
-cholestane-3![]() -yl acetate (1b) (X=O, R=C8H17),5 respectively) upon heating in acetic acid, or under
acid-catalyzed conditions, undergo to a new type of intramolecular rearrangement to give the
corresponding 1',2'-unsaturated (5R,9R)- and (5R,9S)-spiro-
-yl acetate (1b) (X=O, R=C8H17),5 respectively) upon heating in acetic acid, or under
acid-catalyzed conditions, undergo to a new type of intramolecular rearrangement to give the
corresponding 1',2'-unsaturated (5R,9R)- and (5R,9S)-spiro-![]() -lactones 4a,b and 5a,b,
respectively (Scheme 1 and Table 1).
-lactones 4a,b and 5a,b,
respectively (Scheme 1 and Table 1).
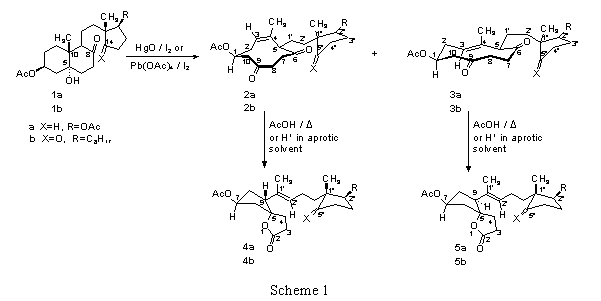
Table 1 Products of the thermal and acid-catalyzed reaction of the (Z)- and (E)-cyclodecenediones 2a, 3a and 2b, 3b
| Substrate | Conditions | Producta |
| / AcOH, r.t.time (h) | yield (%)b | |
| 2a | 4 | 4a (66.0) |
| 3a | 4 | 5a (59.3) |
| 2b | 4.5 | 4b (47.6) |
| 3b | 14 | 5b (9.5) |
| HI / CCl4, r.t. time (h) | ||
| 2a | 2 | 4a (77.4) |
| 3a | 4 | 5a (53.4) |
| 2b | 1 | 4b (52.4) |
| 3b | 2.5 | 5b (54.3) |
| HClO4 / acetone, r.t. time(h) | ||
| 2a | 0.5 | 4a (48.2) |
| 3a | 1 | 5a (62.5) |
a The residue was a complex mixture. bThe results taken from Ref.2.
This transformation takes place by participation of three bonds of the respective (Z)- and (E)-cyclodecenedione rings, i.e., i) the olefinic C(3)=C(4) bond; ii) the keto carbonyl C(9)=O bond;
and iii) the single C(5)-C(6) bond. These bonds are cleared, while three new bonds are formed,
i.e., i) a ![]() -bond between C(3) and C(9); ii) an ether bond between the C(9) carbonyl O
atom and C(6) carbonyl C atom; and iii) a
-bond between C(3) and C(9); ii) an ether bond between the C(9) carbonyl O
atom and C(6) carbonyl C atom; and iii) a ![]() -bond between C(4) and C(5).
-bond between C(4) and C(5).
Therefore, formally, the above rearrangement could be considered as an "ene-type" reaction. However, the results from Table 1 suggest that, at least when performed in the presence of an acid, it is initiated by protonation, most probably at the C(9) or C(6) oxygen, to give the corresponding oxonium ions of type A and B, respectively (see Figures 1 and 2). Although for both species an intramolecular rearrangement to the spirolactones can be envisaged, these processes, depending on the site of protonation, should proceed by two different reaction courses.
In order to elucidate which of these possibilities could be a more reliable pathway to the spiro-![]() -lactone structures 4 (from the (Z)-isomers) and 5 (from the (E)-isomers), the
semiempirical MNDO-AM1 molecular-orbital calculations have been done.
-lactone structures 4 (from the (Z)-isomers) and 5 (from the (E)-isomers), the
semiempirical MNDO-AM1 molecular-orbital calculations have been done.
Method of calculation
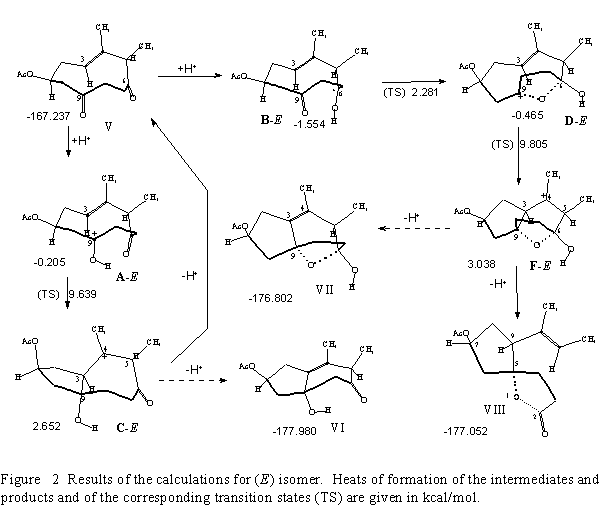
As model compounds the stereoisomeric (1S,5R,Z)- and (1S,5R,E)-4,5-dimethyl-6,9-dioxocyclodec-3-en-1-yl acetates (I and V, Figures 1 and 2), were selected.
The geometries and charge distributions of the molecules were determined by the AM1 method
(using a MOPAC package, Version 7.01)6, employing full geometry optimization and imposing
no a priori symmetry constraints. The MNDO-AM1 method has proven to be fairly accurate for
calculation of molecular properties in various species7. All transition states were proven by
vibrational analysis showing single negative vibration. Simulation of intrinsic reaction
coordinates, starting from TS gives corresponding reactant and product structures.
Results and discussion
(Z)-isomer, I
Reaction intermediates and products of the simulated acid-catalyzed rearrangement of the (Z) compound I are shown in the Figure 1.
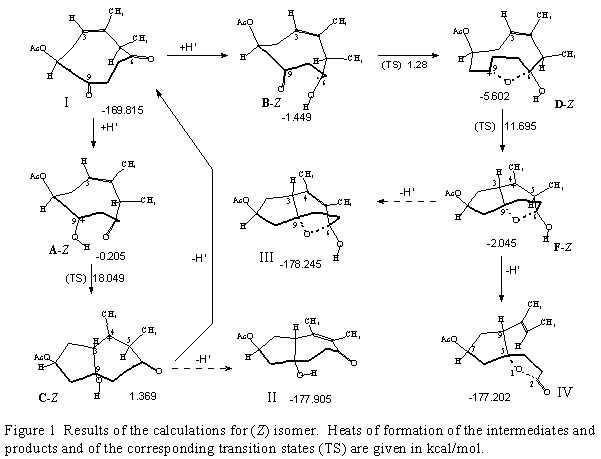
Protonation of the C(9) oxygen produces the less stable (with respect to the C(6)=O protonated
species B-Z) intermediate cation A-Z (0.205 kcal/mol versus 1.449 kcal/mol). In the next step
intramolecular C(3)C(9) cyclization in A-Z, due to the large separation between the reacting
centers, proceeds through an energy rich transition state (TS = 18.049 kcal/mol), to give the C(4)
carbocationic intermediate C-Z. The energy partitioning analysis indicates that the weakest bond
in C-Z is its C(5)H bond, but, deprotonation involving this position would give product II.
Another possibility, i.e., deprotonation from the 9![]() -hydroxyl, gives back the starting
compound I.
-hydroxyl, gives back the starting
compound I.
In the alternative reaction path the more stable intermediate cation B-Z (protonated at C(6)
oxygen), is transformed, practically without activation enthalpy to the more stable C(9)-carbocationic hemiacetale intermediate D-Z. In this species the internuclear separation between
C(3) and C(9) atoms is 3.069 , which is much shorter than the sum of the Van der Waals radii
of these atoms (3.7 ). Therefore, C(3)C(9) cyclization in D-Z to the C(4)-centered
carbocation F-Z proceeds with a considerably lower energy (TS = 11.69 kcal/mol) than C(3)-C(9) cyclization in A-Z (TS = 18.05 kcal/mol). The energy partitioning analysis in F-Z reveals
that C(5)H is the weakest bond in this species, the deprotonation of which would afford product
III. However, deprotonation from the 6![]() -hydroxyl spontaneously leads to the
experimentally found spiro-
-hydroxyl spontaneously leads to the
experimentally found spiro-![]() -lactone structure of type IV.
-lactone structure of type IV.
(E)-isomer, V
Reaction intermediates and products of the transformation of the (E)-compound identified by AM1 calculations, are shown in Figure 2. The results obtained are analogous to those of the (Z)-isomer.
 .
.
Protonation of oxygen atoms at C(9) and C(6), respectively, yields the corresponding oxonium
ions A-E and B-E, the latter being more stable than the former (1.554 kcal/mol versus 0.205
kcal/mol). Cyclization of A-E by formation of the C(3)C(9) bond affords cationic intermediate
C-E (TS = 9.639 kcal/mol), which can be stabilized either by elimination of the most labile
C(3)H proton to give compound VI or by deprotonation from the 9![]() -hydroxyl to recover
the starting product V.
-hydroxyl to recover
the starting product V.
On the other hand, B-E protonated at C(6) oxygen, is rearranged, via the hemiketale species D-E, to the intermediate F-E. (In order to check thee alternative reaction sequence: first formation
of CC, and then of CO bond, the C(3)C(9) distance in structure B-E was treated as reaction
coordinate. On this route the structure D-E is formed first.) The C(3)C(9) distance in
intermediate D-E is only 2.514 , and, consistently, the transition state for conversion to F-E has
low energy (TS = 9.805 kcal/mol). In this intermediate deprotonation involving its weakest
(C(3)H) bond results in formation of the ![]() 3-unsaturated compounds VII. However, upon
deprotonation from the C(6) hydroxyl the spiro-
3-unsaturated compounds VII. However, upon
deprotonation from the C(6) hydroxyl the spiro-![]() -lactone structure VIII is
spontaneously formed.
-lactone structure VIII is
spontaneously formed.
The above calculations indicate that intermediates C and F should be preferentially stabilized by C-deprotonation, involving hydrogen in the vicinity of the carbocationic site, thus producing compounds II and III in the (Z)-series and compounds VI and VII in the (E)-series. However, the structural analogoues of these compounds were not found among reaction products of the steroid (Z)- and (E)-cyclodecendione derivatives of type 2 and 3. As can be seen from Scheme 1, experimentally only products of O-deprotonation were identified.(3) This is consistent with the Marcus model of proton transfer, which predict much higher rate for proton exchange from the O- and N-bases, in comparison to the C-bases.6 This could be rationalized by higher positive charge at a hydrogen atom bonded to the more electronegative atom, rather than by strengths of the respective bonds. In addition, the structures IV and VIII have more internal rotational degrees of freedom giving rise to the more positive entropy of formation, making their formation thermodynamically preferable.
In both calculated reaction schemes is evident that the more convenient reaction path to the spiro-![]() -lactone rings IV and VIII, respectively, involves protonation at the C(6) carbonyl
oxygen in the corresponding cyclodecenediones I and V giving the intermediate cation B,
followed by isomerization to the intermediate cation F. After the removal of hydrogen from the
OH group, a spirolactone is spontaneously formed. Furthermore, calculations predict that
spirolactones formed from (Z)- and (E)- substrates should differ in the stereochemistry at the
C(9) carbon, exactly as found experimentally.
-lactone rings IV and VIII, respectively, involves protonation at the C(6) carbonyl
oxygen in the corresponding cyclodecenediones I and V giving the intermediate cation B,
followed by isomerization to the intermediate cation F. After the removal of hydrogen from the
OH group, a spirolactone is spontaneously formed. Furthermore, calculations predict that
spirolactones formed from (Z)- and (E)- substrates should differ in the stereochemistry at the
C(9) carbon, exactly as found experimentally.
Experimentally found higher reactivity of (Z) isomers, In the acid catalyzed reaction, exemplified
by shorter reaction times (see Table 1), is in accord with more convenient conformation of (Z)
isomer of intermediate B for intramolecular cyclization. The distance between oxygen and
carbon atoms that form an ether bond in the next reaction step is shorter in (Z) isomer (2.729 )
than in (E) isomer (2.744 ). Both distances are considerably smaller than the corresponding
Van der Waals distance (3.20 ).
Acknowledgements
We thank the Serbian Academy of Sciences and Arts and the Serbian Ministry of Sciences and
Technology for financial support.
References
[1] LJ. LORENC, L. BONDARENKO-GHEORGHIU, N. KRSTIC, H. FUHRER, J. KALVODA , M. LJ. MIHAILOVIC, Helv. Chem. Acta, 78, 891 (1995).
[2] LJ. LORENC, L. BONDARENKO-GHEORGHIU, V. PAVLOVIC, N. KRSTIC, M. LJ. MIHAILOVIC, J. Serb. Chem. Soc., 63, 833 (1998).
[3] M.LJ. MIHAILOVIC, LJ. LORENC, L. BONDARENKO, B. TINANT, J-P. DECLERCQ, M. VAN MEERSSCHE, Tetrahedron, 42, 189 (1986).
[4] LJ. LORENC, L. BONDARENKO, V. PAVLOVIC, H. FUHRER, G. RIHS, J. KALVODA, M.LJ. MIHAILOVIC, Helv. Chem. Acta, 72, 608 (1989).
[5] M. LJ. MIHAILOVIC, V. D. PAVLOVIC, L. G. BONRADENKO-GHEORGHIU, N. M. KRSTIC, M. DABOVIC, LJ. B. LORENC, J. Serb. Chem. Soc., 61, 941 (1996).
[6] J. J. P. STEWART, J. Comput. Chemistry, 2, 221 (1989).
[7] T. WYTTENBACH, G. VONHELDEN, M. T. BOWERS, J. Am. Chem. Soc., 118, 8355 (1996); H. BOCK, S. NICK, W. SEITZ, C. NATHER, J. W. BATS, Z. Naturforsch. B, 51, 153 (1996); G. H. PESLHERBE, H. B. WANG, W.L. HASE, J. Am. Chem. Soc., 118, 2257 (1996);W. P. HU, D. G. TRUHLAR, J. Am. Chem. Soc., 118, 860 (1996); I. TABAKOVIC, E. GUNIC, I. JURANIC: J. Org. Chem., 62, 947 (1997).
[8] R. A. MARCUS, J. Phys. Chem., 72, 891 (1968); A. O. COHEN, R. A. MARCUS, ibid.,
p.4249; J. R. MURDOCH, J. Am. Chem. Soc., 94, 4410 (1972); W. ALBERY, A. Rev.
Phys. Chem., 31, 227 (1980).
1. Work supported in part by the Ministry of Science and Technology of the Republic of Serbia.
2. To whom correspondence should be addressed.
3. The C-deprotonated compounds of type II and III in the (Z)-series, and of type VI and VII in the (E)-series, if formed at all, could be present as components of the respective complex mixtures (see Table 1), however, in very low yields.